Code & Data
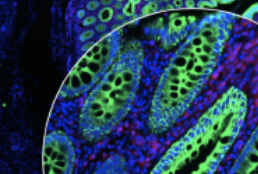
Scope2Screen is a scalable software system for focus+context exploration and annotation of whole-slide, high-plex, tissue images.
Scope2Screen: focus+context techniques for pathology tumor assessment in multivariate image data. Jessup J, Krueger R, Warchol S, Hoffer J, Muhlich J, Ritch CC, Gaglia G, Coy S, Chen Y, Lin J, Santagata S, Sorger PK, Pfister H. IEEE Trans Vis Comput Graph. 2022 Jan;28(1):259-269. doi: 10.1109/TVCG.2021.3114786. PMID: 34606456. PMCID: PMC8805697.
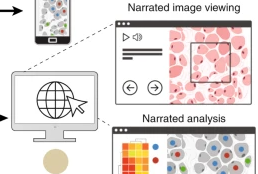
Minerva is a suite of software tools for tissue atlases and digital pathology that enables interactive viewing and fast sharing of large image data. Development of Minerva is led by Jeremy Muhlich and John Hoffer.
Minerva: a light-weight, narrative image browser for multiplexed tissue images.mHoffer J, Rashid R, Muhlich JL, Chen Y-A, Russell DPW, Ruokonen J, Krueger R, Pfister H, Santagata S, Sorger PK. J Open Source Softw. 2020 Oct 15;5(54):2579. doi: 10.21105/joss.02579. PMID: 33768192. PMCID: PMC7989801.
Narrative online guides for the interpretation of digital-pathology images and tissue-atlas data. Rashid R, Chen YA, Hoffer J, Muhlich JL, Lin JR, Krueger R, Pfister H, Mitchell R, Santagata S, Sorger PK. Nat Biomed Eng. 2021 Nov 8. doi: 10.1038/s41551-021-00789-8. Epub ahead of print. PMID: 34750536. PMCID: PMC9079188.
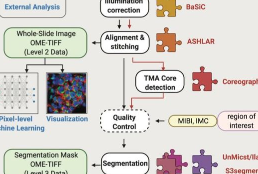
MCMICRO is the end-to-end processing pipeline for multiplexed whole tissue imaging and tissue microarrays. It comprises stitching and registration, segmentation, and single-cell feature extraction. Development of mcmicro is led by Artem Sokolov and Denis Schapiro.
MCMICRO: a scalable, modular image-processing pipeline for multiplexed tissue imaging. Schapiro D, Sokolov A, Yapp C, Chen YA, Muhlich JL, Hess J, Creason AL, Nirmal AJ, Baker GJ, Nariya MK, Lin JR, Maliga Z, Jacobson CA, Hodgman MW, Ruokonen J, Farhi SL, Abbondanza D, McKinley ET, Persson D, Betts C, Sivagnanam S, Regev A, Goecks J, Coffey RJ, Coussens LM, Santagata S, Sorger PK. Nat Methods. 2022 Mar;19(3):311-315. doi: 10.1038/s41592-021-01308-y. PMID: 34824477. PMCID: PMC8916956.
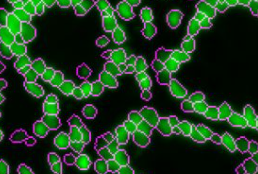
UnMicst uses the popular UNet architecture to semantically segment an image of nuclei stained with DAPI and/or lamin. UnMicst stands for UNet Model for Identifying Cells and Segmenting Tissue. Version 1 is an early model trained on a small number of annotations. Version 2 has been trained on lung, fibroblast, tonsil, glioblastoma, small intestine, and colon tissue. Development of UnMicst is led by Clarence Yapp.
UnMICST: Deep learning with real augmentation for robust segmentation of highly multiplexed images of human tissues. Yapp C, Novikov E, Jang WD, Vallius T, Chen YA, Cicconet M, Maliga Z, Jacobson CA, Wei D, Santagata S, Pfister H, Sorger PK. Commun Biol. 2022 Nov 18;5(1):1263. doi: 10.1038/s42003-022-04076-3. PMID: 36400937. PMCID: PMC9674686.
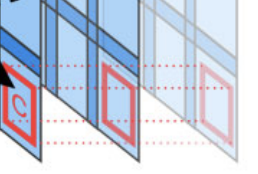
ASHLAR (Alignment by Simultaneous Harmonization of Layer/Adjacency Registration) is an open source Python package that stiches together successive microscopy image tiles to generate a single, seamless image. ASHLAR also registers images from different fluorescent channels at a high level of accuracy.
Stitching and registering highly multiplexed whole slide images of tissues and tumors using ASHLAR. Muhlich JL, Chen YA, Yapp C, Russell D, Santagata S, Sorger PK. Bioinformatics. 2022 Aug 16;38(19):4613–21. doi: 10.1093/bioinformatics/btac544. Epub ahead of print. PMID: 35972352. PMCID: PMC9525007.
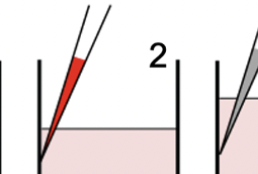
Reliable high-throughput imaging of cells grown in multi-plate wells is complicated by loss of cells during staining and wash steps. The dye drop method uses a set of incrementally more dense solutions to prevent cell loss. Dye Drop software consists of Python tools for determining the viability and cell cycle states of cells before and after drug treatment.
Multiplexed and reproducible high content screening of live and fixed cells using Dye Drop. Mills CE, Subramanian K, Hafner M, Niepel M, Gerosa L, Chung M, Victor C, Gaudio B, Yapp C, Nirmal AJ, Clark N, Sorger PK. Nat Commun. 2022 Nov 14;13(1):6918. doi: 10.1038/s41467-022-34536-7. PMID: 36376301; PMCID: PMC9663587.
The DRIAD tool evaluates gene sets for their ability to predict the progression of Alzheimer’s disease (AD). It utilizes expression data from post-mortem samples of AD patients collected by the AMP-AD consortium.
Machine learning identifies candidates for drug repurposing in Alzheimer’s disease. Rodriguez S, Hug C, Todorov P, Moret N, Boswell SA, Evans K, Zhou G, Johnson NT, Hyman BT, Sorger PK, Albers MW, Sokolov A. Nat Commun. 2021 Feb 15;12(1):1033. doi: 10.1038/s41467-021-21330-0. PMID: 33589615. PMCID: PMC7884393.
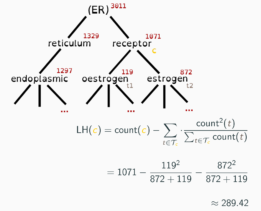
Adeft (Acromine based Disambiguation of Entities From Text context) is a utility for building models to disambiguate acronyms and other abbreviations of biological terms in the scientific literature. It makes use of an implementation of the Acromine algorithm developed by the NaCTeM at the University of Manchester to identify possible longform expansions for shortforms in a text corpus. It allows users to build disambiguation models to disambiguate shortforms based on their text context. A growing number of pretrained disambiguation models are publicly available to download through adeft.
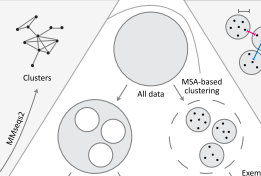
ProteinNet is a standardized data set for machine learning of protein structure. It provides protein sequences, structures (secondary and tertiary), multiple sequence alignments (MSAs), position-specific scoring matrices (PSSMs), and standardized training / validation / test splits. ProteinNet builds on the biennial CASP assessments, which carry out blind predictions of recently solved but publicly unavailable protein structures, to provide test sets that push the frontiers of computational methodology. It is organized as a series of data sets, spanning CASP 7 through 12 (covering a ten-year period), to provide a range of data set sizes that enable assessment of new methods in relatively data poor and data rich regimes.
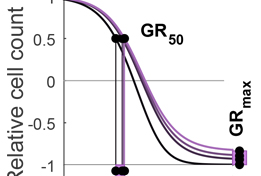
The Growth Rate inhibition (GR) Calculator is an open source set of Python, R and on-line tools for quantifying the responses of cancer cells to drugs in a manner that corrects for the confounding effects of variable cell proliferation rates. Response metrics computed from GR data include GR50 and GRmax and are direct analogues of familiar IC50 and Emax response measures.
Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Hafner M, Niepel M, Chung M, Sorger PK. Nat Methods. 2016. 13(6):521-7. doi: 10.1038/nmeth.3853.PMID: 27135972.
Alternative drug sensitivity metrics improve preclinical cancer pharmacogenomics. Hafner M, Niepel M, Sorger PK. Nat Biotechnol. 2017. 35(6):500-502. doi: 10.1038/nbt.3882. PMID: 28591115.
-
- The CCSP provides all its content, tools, and data on an “as is” basis, without warranty or representation of any kind, express or implied. If you use code, data or other content from this website, you must accept the terms on this page.
- We reserve the right to modify these terms at any time. Unless otherwise specified, text, images and data on the site are provided under a Creative Commons Attribution ShareAlike (CC BY-SA) license. Scientific manuscripts carry the licenses of their publishers.

- The CCSP aims to provide timely public access to all relevant data, software, and tools in accordance with data release policy of the NCI Cancer System Biology Consortium.
- Note that data released prepublication is potentially subject to confounders, batch effects and other errors which may not have been identified or controlled. While the CCSP works hard to ensure the reproducibility of its results, use of pre-publication data entails extra risks.
- Specialized reagents (e.g. cell lines, plasmids etc.) are generally made available via standard repositories (e.g. Addgene). In other cases, contact us at the address below. Reagents that are not in repositories will usually require an MTA prior to distribution. We do not redistribute materials that originated with commercial vendors or outside research groups; we will not respond to requests about such materials.
- Users are expected to acknowledge the following in all oral or written presentations, disclosures, or publications of the data or analyses provided by the CCSP.
- For unpublished data found on this site:
The Harvard Medical School CCSP and the funding source that supported the work: NIH grant U54-CA225088 (e.g. “[x] data were provide by the Harvard Medical School Center for Cancer Systems Pharmacology, funded by NIH grant U54-CA225088.”) - For published data:
The normal rules of scientific citation apply. Whenever feasible we use open source licenses for our manuscripts and data. Data are also routinely deposited in GEO, PRIDE, and other repositories. Large-scale imaging data remain hard to share; please contact us if you need access. - For software:
Software is generally released under an MIT Open Source license. See https://github.com/sorgerlab for details.
- For unpublished data found on this site:
If you have specific questions or comments about HMS CCSP data, software, and tools, contact us at Alyce_Chen@hms.harvard.edu.